La loi des gaz parfaits est une loi approchée qui ne représente correctement
le comportement des gaz qu'aux faibles pressions.
Dans un diagramme d'Amagat (PV,P) les isothermes d'un
gaz parfait sont des droites horizontales.
Les mesures effectuées sur les gaz réels permettent de tracer le réseau
des isothermes des gaz réels, par exemple ici l'azote.
Vers les basses températures, les courbes se déforment jusqu'à présenter
un point d'inflexion. L'isotherme correspondante est appelée isotherme
critique. En dessous de cette température (-147 °C pour l'azote),
le gaz peut être liquéfié.
La température pour laquelle la pente de l'isotherme aux basses pressions
est nulle est appelée température de Mariotte (50 °C pour l'azote).
De trés nombreuses équations d'état ont été proposées pour rendre
compte du comportement réel des fluides. La plus connue, et la plus
simple, est l'équation de Van der Waals:
⎛
⎜
⎜
⎝
P+
a
V2
⎞
⎟
⎟
⎠
⎛
⎝
V−b
⎞
⎠
=r.T
Malheureusement, si cette équation permet d'obtenir un réseau d'isothermes
ayant bien l'allure d'un reseau réel, elle ne permet pas d'obtenir
des résultats numériques acceptables dans un large domaine de pressions
et de températures. Elle a surtout une importance historique car c'est
la première à permettre de représenter les deux phases gazeuse et
liquide en équilibre. De plus elle est obtenue à partir un raisonnement
moléculaire très pertinent. La raison pour laquelle elle est peu précise
est qu'elle ne prend pas en compte le détail des forces d'attraction
et de répulsion agissant au niveau moléculaire. Aucune équation simple
n'est en mesure de donner des résultats précis pour tous les fluides
quels que soient la température et la pression.
La plupart des équations d'état utilisées dans l'industrie sont des
extensions de l'équation de van der Waals. De plus, toutes les équations
d'état ont en commun de tendre vers la loi des gaz parfaits pour les
faibles pressions.
Les calculs sont complexes avec la plupart de ces équations d'état
et pour s'éviter des calculs fastidieux, notamment en présence de
changements d'état, on est amené à utiliser des diagrammes fournissant
des solutions graphiques approximatives mais quasi-immédiates. Par
ailleurs, le developpement de l'informatique permet aujourd'hui d'obtenir
des solutions rapides à l'aide de logiciels appropriés.
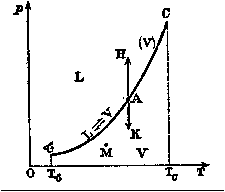
Dans un tube maintenu à température constante, on enferme du CO2
puis on fait varier la pression et le volume en poussant sur le piston.
Lorsqu'on atteint une certaine pression Pv, une première goutte
de liquide apparait. Si l'on continue à enfoncer le piston, la masse
de liquide produite augmente mais la pression reste constante
tandis que le volume occupé par le mélange diminue.
Lorsqu'il n'y a plus que du liquide, la pression augmente très rapidement
tandis que le volume diminue très lentement. Si l'on répéte l'expérience
pour d'autres températures, on obtient le réseau d'isothermes ci-dessous.
Au delà de la température critique, la liquéfaction n'est plus possible
(31°C pour CO2).
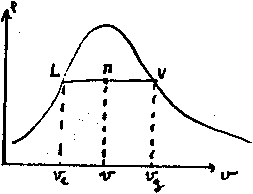
La courbe (S) est appelée courbe de saturation, elle représente l'état
du liquide pur (à gauche du point critique C) et de la vapeur saturée
pure (à droite du point C). Les points situés en dessous de la courbe
correspondent à un mélange de liquide et de vapeur en équilibre.
Pour un équilibre liquide-vapeur, la pression et la température sont
liées par une fonction Ps=f(Ts); c'est un système
monovariant (c'est à dire à un seul degré de liberté).
La courbe de pression de vapeur est limitée par le point critique
C et le point triple T, point de coexistence des trois phases (vapeur,
liquide, solide). La pente de la courbe n'est pas infinie en C, et
différentes formules peuvent représenter cete courbe:
- formule de Rankine: Log(Ps)=A−B/T
- formule de Duperray, pour l'eau entre 100 et 200 °C: Ps=(t/100)4,
avec Ps en bars et t en °C.
C'est la variation d'enthalpie d'une masse unitaire de liquide saturant
qui se vaporise totalement à pression constante, et température constante
évidemment.
L=Hv−Hl=Uv−Ul+P
⎛
⎝
Vv−Vl
⎞
⎠
=Qp
C'est la quantité de chaleur qu'il faut fournir, à pression constante,
pour vaporiser le liquide, L dépend de la température. Les mesures
effectuées donnent une courbe de la forme ci-contre, exemple pour
l'eau, L tend vers 0 pour la température critique.
La formule qui suit peut être démontrée à partir des deux principes.
Elle n'est pas une formule approchée mais une formule thermodynamiquement
exacte. Elle relie la chaleur latente à la pente de la courbe de saturation.
De plus des formules analogues existent n'importe quel équilibre entre
phases d'un corps pur. (par exemple l'équilibre liquide solide)
L=T
⎛
⎝
vg−vl
⎞
⎠
dPs
dT
avec vg et vl volume massique de la vapeur et du liquide
saturant.